
This web page was produced as an assignment for Genetics 564, and undergraduate course at UW-Madison.
What is phylogeny?
A phylogeny is a tool that can be used to visualize the relationships between different species. Originally, phylogenies were based solely upon a species appearance and anatomy, with no quantitative way to determine their accuracy [1]. With advances in DNA and protein sequences, scientists are now able to use information hidden in an organisms genetic code to determine their evolutionary history and create a phylogeny from this information [1]. Now, there are a variety of tools and methods available to researchers to generate phylogenies for organisms, genes, or proteins that they wish to study [2].
Shown below are two phylogenetic trees made of the RAG1 gene using different methods to generate the tree. The first tree is a neighbor joining tree. In a neighbor joining tree, organisms are paired in a way to make the shortest distance between organisms [3]. In contrast, the average distance tree looks at the sequences of the organisms present and tries to minimize the difference in genomes among organisms.
Figures 1 and 3 below are made using the percent identity method. In this method, the algorithm looks at the number of equivalent amino acids at each location and compares it to number of possible amino acids at each location. This is used to calculate the percent similarity between organisms and filter them into an appropriate tree. Another method of building trees is with an algorithm called Blosum. This algorithm factors in the likelihood of one amino acid being substituted in for another [5]. Figures 2 and 4 are made with this method.
Shown below are two phylogenetic trees made of the RAG1 gene using different methods to generate the tree. The first tree is a neighbor joining tree. In a neighbor joining tree, organisms are paired in a way to make the shortest distance between organisms [3]. In contrast, the average distance tree looks at the sequences of the organisms present and tries to minimize the difference in genomes among organisms.
Figures 1 and 3 below are made using the percent identity method. In this method, the algorithm looks at the number of equivalent amino acids at each location and compares it to number of possible amino acids at each location. This is used to calculate the percent similarity between organisms and filter them into an appropriate tree. Another method of building trees is with an algorithm called Blosum. This algorithm factors in the likelihood of one amino acid being substituted in for another [5]. Figures 2 and 4 are made with this method.
Neighbor joining tree
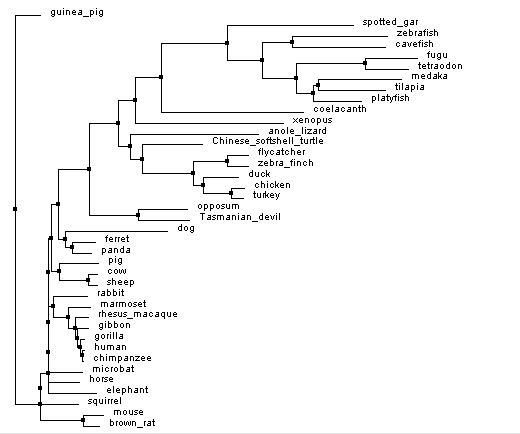
Figure 1: Neighbor joining tree by percent identity. Generated with Clustal Omega, viewed with Jalview. Click image to enlarge
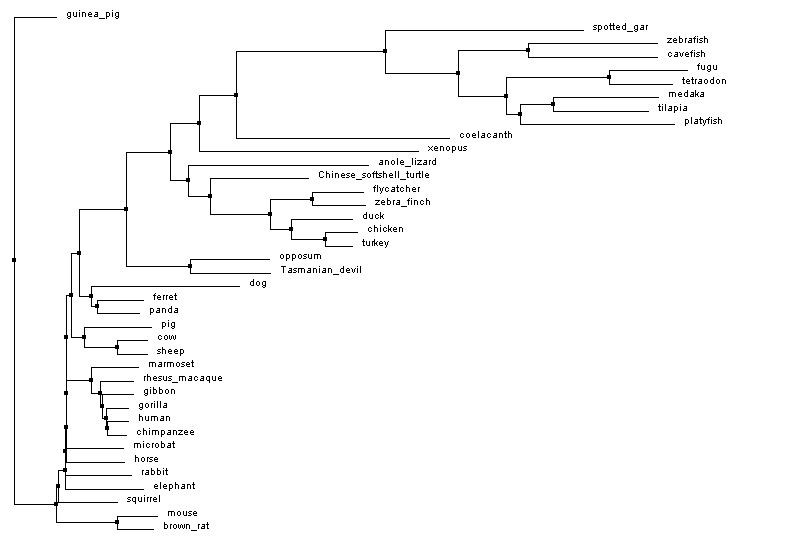
Figure 2: Neighbor joining tree by Blosum. Generated with Clustal Omega, viewed with Jalview. Click image to enlarge
Average distance tree
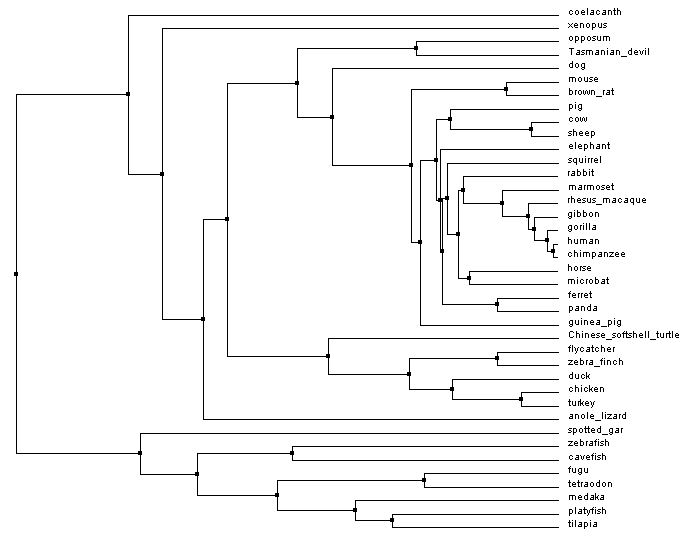
Figure 3: Average distance tree by percent identity. Generated with Clustal Omega, viewed with Jalview. Click to enlarge
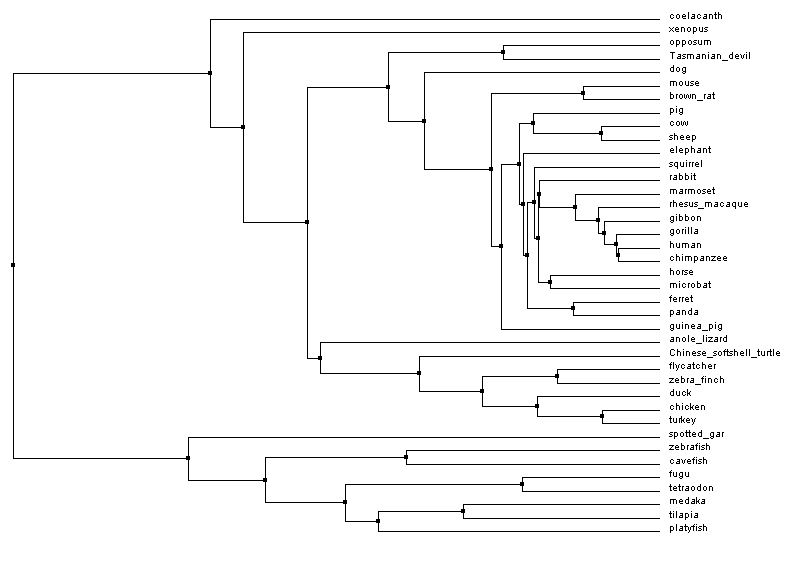
Figure 4: Average distance tree by Blosum. Generated with Clustal Omega, viewed with Jalview
Discussion
The different phylogenies generated by different methods are very similar for RAG1. There is also a high degree of conservation among different organisms, especially animals of the same class. This suggests an important role in the conservation of the protein.
References
1. phylogeny. (2015). In Encyclopædia Britannica. Retrieved from http://www.britannica.com/EBchecked/topic/458573/phylogeny
2. Yang, Z. and Rannala, B. Molecular phylogenetics: principles and practice. Nature reviews 13 (2012) 303-314
3. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987 Jul;4(4):406-25. PubMed PMID: 3447015.
4. "Calculation of trees from alignments" Jalview. <http://www.jalview.org/help/html/calculations/tree.html>
5. "Substitution matrices available in Jalview" Jalview <http://www.jalview.org/help/html/calculations/scorematrices.html>
Image References
Header image: http://commons.wikimedia.org/wiki/File:Phylogenetic_Tree_of_Life.png
2. Yang, Z. and Rannala, B. Molecular phylogenetics: principles and practice. Nature reviews 13 (2012) 303-314
3. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987 Jul;4(4):406-25. PubMed PMID: 3447015.
4. "Calculation of trees from alignments" Jalview. <http://www.jalview.org/help/html/calculations/tree.html>
5. "Substitution matrices available in Jalview" Jalview <http://www.jalview.org/help/html/calculations/scorematrices.html>
Image References
Header image: http://commons.wikimedia.org/wiki/File:Phylogenetic_Tree_of_Life.png
